files - research summary
Stec Research Summary
My lab is focused on understanding the physiological role of the heme oxygenase (HO) system in the regulation of the cardiovascular system and metabolism. HO is the enzyme responsible for the breakdown of heme into free iron (Fe2+), carbon monoxide (CO), and biliverdin. Biliverdin is then rapidly reduced to bilirubin by the enzyme biliverdin reductase (BVR). My lab is interested not only in the enzymes HO and BVR but in the metabolites, CO and bilirubin.
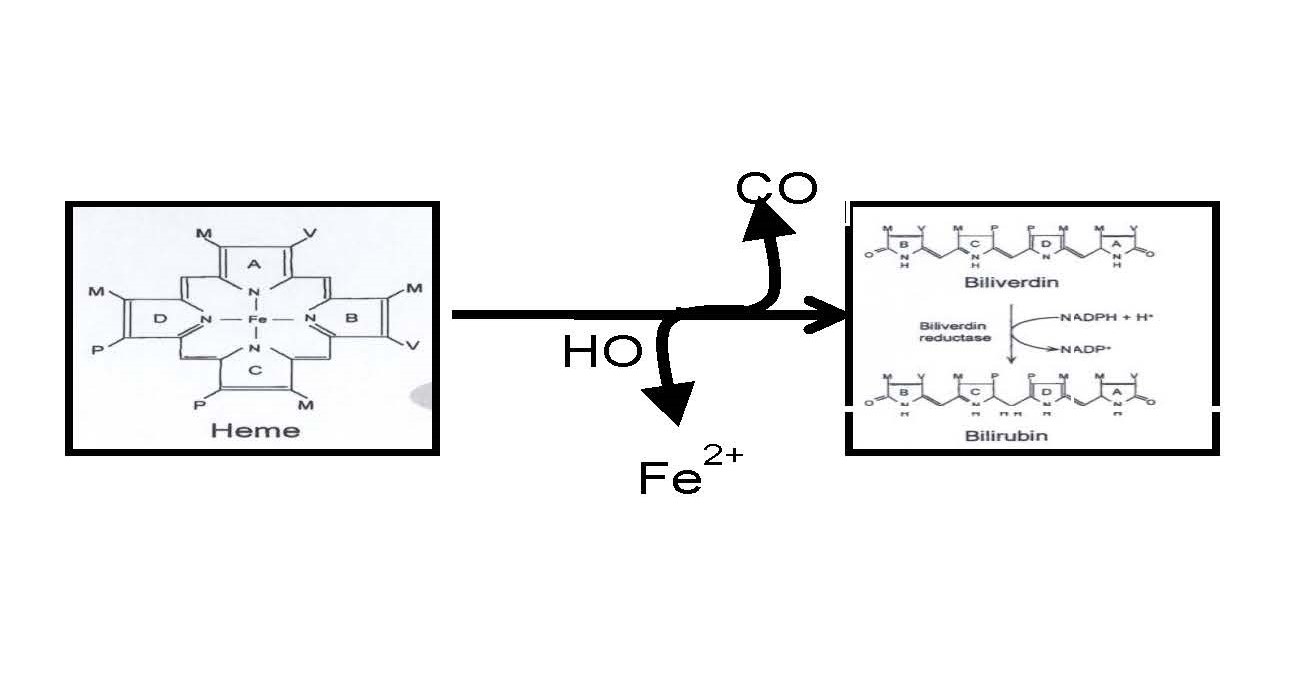
One of the areas in which we have been working is the on the role of HO system in obesity and its related cardiovascular diseases. We have previously demonstrated that chronic induction of HO-1 lowers body weight, increases oxygen consumption, increases carbon dioxide (CO2) production, increases heat production and activity, and increases plasma adiponectin levels in two mouse models of obesity due to melanocortin-4 receptor deficiency. We have recently demonstrated a sex-specific effect of loss of HO-1 specifically from adipocytes. Female mice have more metabolic complications from loss of adipocyte HO-1 than males despite no changes in overall body weight.
Our lab is also focused on the role of increased levels of plasma bilirubin, a metabolite of HO, in the prevention of cardiovascular disease. Many large scale patient population studies have demonstrated a link between increased levels of plasma bilirubin (between 50% to 2 fold increase) and the prevention of cardiovascular diseases including stroke, heart disease, hypertension, obesity, and hepatic steatosis. However, despite these clinical correlations, the mechanism by which moderate increases in plasma bilirubin protect against cardiovascular disease is unknown. Bilirubin has long been appreciated as a major antioxidant in the body but apart from this no other function of bilirubin is known. Recently, our lab in collaboration with the lab of Dr. Terry D. Hinds, Jr. at the University of Kentucky School of Medicine have identified bilirubin as a signaling molecule capable of activating nuclear receptors such as peroxisome proliferator-activated receptor-a (PPARa). This is the first new physiological function described for bilirubin in the last 30 years. We have also shown in a recent study that mice with a genetic form of human hyperbilirubinemia (Gilbert’s polymorphism) are protected against dietary-induced obesity, hyperglycemia, and hepatic steatosis in part through bilirubin stimulating PPARa in the liver.
We are currently performing studies to determine the role of bilirubin signaling through PPARa in the anti-steatotic and anti-diabetic actions of moderate hyperbilirubinemia. Many clinical and population studies have determined that low levels of bilirubin are associated with cardiovascular and metabolic diseases including obesity, diabetes, and non-alcoholic fatty liver disease (NAFLD). Bilirubin is very insoluble in water so it is not an ideal candidate for drug treatment. However, we have utilized a soluble form of bilirubin nanoparticles (PEG-BR) for the treatment of dietary obesity-induced NAFLD and insulin resistance. We are currently examining more effective ways to deliver this compound for the treatment of NAFLD. 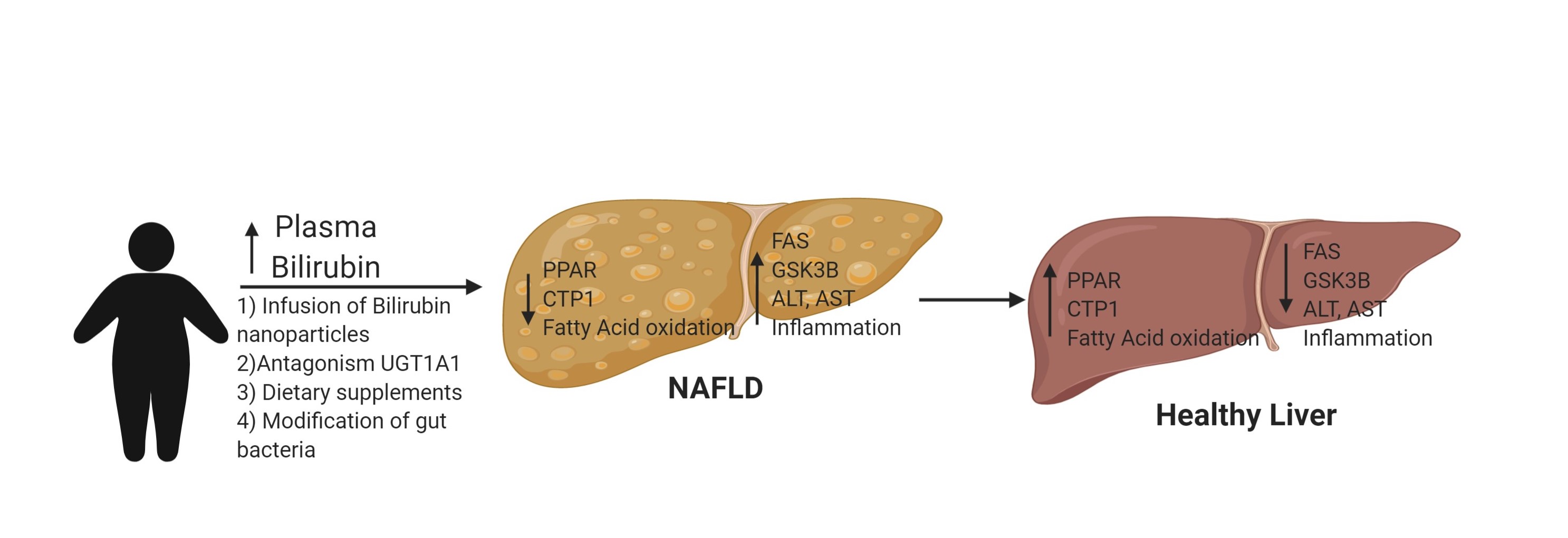
Biliverdin reductase (BVR) is the enzyme responsible for the reduction of biliverdin to bilirubin. It can also act as a signaling molecular through its interactions with the Akt and protein kinase C pathways as well as acting as a regulator of transcription. In order to understand more about the physiological role of the BVRA isoform, we have developed a novel conditional knockout out mouse model of BVRA. We recently reported along with Dr. Hinds that hepatocyte specific knockout of BVRA results in exacerbation of high fat diet-induced hepatic steatosis and hyperglycemia. We discovered that BVRA regulates hepatic steatosis in part through its regulation of glycogen synthase kinase-3b (GSK-3b). BVRA increases phosphorylation of GSK-3b which results in its inactivation. However, loss of BVRA increases GSK-3b activity which increases phosphorylation of PPARa at serine 73 which targets it for degradation and lowers the levels of PPARa which promotes hepatic fat storage. We are currently working on the mechanism by which BVRA regulates GSK-3b phosphorylation and the effect of loss of hepatic BVRA on liver injury due to ischemia/reperfusion and chronic alcohol abuse.
Carbon monoxide (CO) is another important metabolite generated by the HO system. CO has long been known as a poisonous gas; however, it also is produced endogenously by the body and has several important functions. Our lab was the first to demonstrate that chronic administration of carbon monoxide releasing molecules (CORMs) prevents and reverses dietary-induced obesity in mice. CO lowers body weight by increasing metabolism though its effect on fat cells. CO is able to convert fat cells from fat storage to fat burning though its effects on several different genes that regulate mitochondrial oxidation of fatty acids. In addition, CO is able to decrease the release of high-mobility group box protein-1 (HMGB-1) from adipocytes. HMGB-1 is a regulator of systemic inflammation which is increased in models of obesity. One major limitation in the development of new anti-obesity drugs is the adverse cardiovascular actions of drugs that increase metabolism to promote weight loss. We have data demonstrating the CORMs lower blood pressure in addition to increasing metabolism to promote weight loss. Thus, CORMs appear to be ideal candidates for effective, safe weight loss drugs. We are currently working on new formulations of CORMs to develop as novel anti-obesity drugs.
One of the tenants of renal physiology is that increases in renal perfusion pressure (blood pressure) result in increased excretion of sodium and water. This phenomenon is known as the renal pressure-natriuresis relationship. Interestingly, in all animal models of hypertension tested, the relationship between increases in renal perfusion pressure and sodium and water excretion is altered so that individuals with hypertension require higher levels of renal perfusion pressure to excrete normal amounts of sodium and water as compared to individuals with normal blood pressure. My lab has been studying the relationship between several hormone systems and the regulation of renal pressure-natriuresis in hypertension. My lab has been interested in the renal mechanisms that mediate the antihypertensive actions of HO-1 induction in the kidney. Our group first reported that kidney specific induction of HO-1 was able to reduce blood pressure in a model of angiotensin (Ang) II-dependent hypertension. We also created a novel transgenic mouse model in which the HO-1 isoform is expressed specifically in thick ascending loop of Henle (TALH) cells in the kidney. We have demonstrated that these mice exhibit a lower blood pressure after infusion of Ang II. Interestingly, these mice exhibit decreased levels of the Na, K, 2Cl (NKCC2) transporter in the kidney. We are currently performing experiments to determine the regulation of the NKCC2 transporter by HO-1 and its metabolites. We are also developing both kidney specific overexpression models in nephron segments such as the proximal tubule, and collecting duct to determine the effects of increased levels of HO-1 in these nephron segments on sodium reabsorption and blood pressure. In addition, we have received mice which contain a conditional knockout allele of HO-1 and are developing mice with kidney tubule segment specific deficiency in HO-1 to understand the role of endogenous HO-1 and its metabolites in the regulation of sodium reabsorption and blood pressure.